Editor’s Note: This is part 3 of a 7 part series on Process Risk Assessments. If you’re just getting started with the series, check out part 1.
Last week we discussed how to use the concepts of ISO 14971 to create a generalized framework for risk management that works across pharma and is compatible with (and leverages the best parts from) medical devices.
This week, we’re going to take that one step further. We will explore how these concepts line up with FMEA/FMECA methodologies.
Let’s review!
Focus on FMEA/FMECA
The QbDVision platform is intended to align with the recommendations outlined in the various ICH guidelines, specifically ICH Q8 to ICH Q12. ICH Q9 specifically discusses the concepts of quality risk management but does not recommend a specific risk methodology for use in the risk assessment of manufacturing processes. However, we find that recent writings by the FDA do recommend the use of FMEA/FMECA for the risk assessment of pharmaceutical manufacturing processes related to their knowledge-aided assessment & structured applications (KASA) initiative. For this reason, the risk assessment features and functionalities available in QbDVision are based on the FMEA/FMECA methodologies. ICH Q9 defines FMEA and FMECA as follows.
FMEA – Failure Mode Effects and Analysis
FMEA provides for an evaluation of potential failure modes for processes and their likely effect on outcomes and/or product performance. Once failure modes are established, risk reduction can be used to eliminate, contain, reduce or control the potential failures. FMEA relies on product and process understanding and methodically breaks down the analysis of complex processes into manageable steps. It is a powerful tool for summarizing the important modes of failure, factors causing these failures and the likely effects of these failures.
FMECA – Failure Mode Effects and Criticality Analysis
FMEA might be extended to incorporate an investigation of the degree of severity of the consequences, their respective probabilities of occurrence, and their detectability, thereby becoming a Failure Mode Effect and Criticality Analysis (FMECA; see IEC 60812). In order for such an analysis to be performed, the product or process specifications should be established. FMECA can identify places where additional preventive actions might be appropriate to minimize risks.
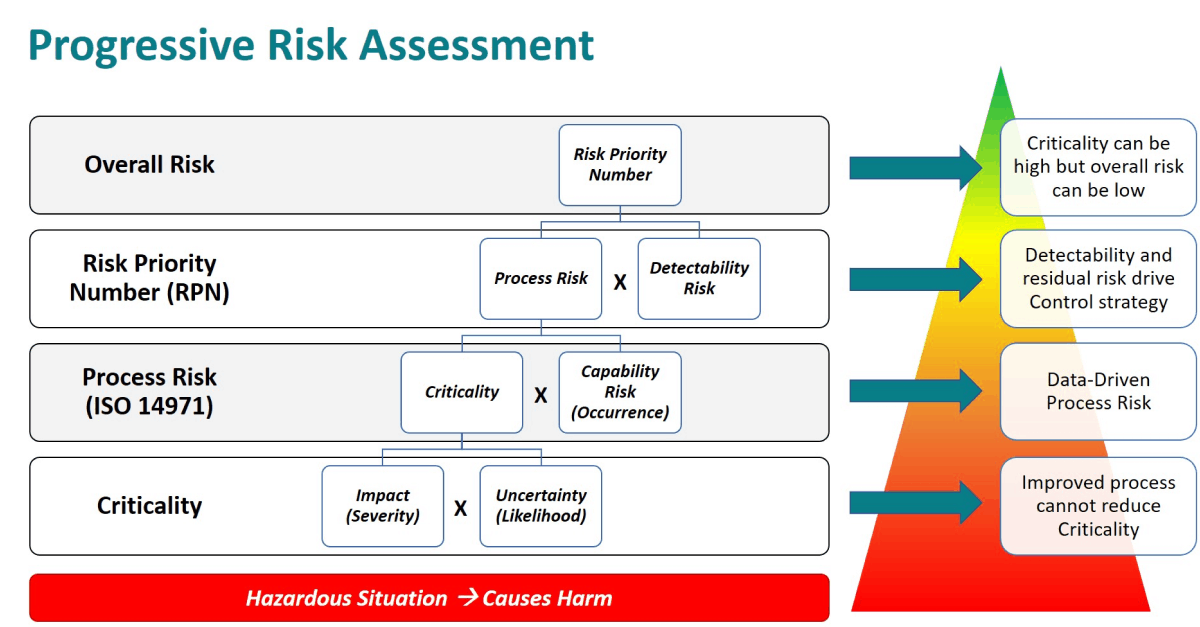
In practice, and in QbDVision, risk assessment is a progressive exercise, as diagrammed last week where the quantitative estimation of each layer should be based on data and/or sound scientific rationale. Risk assessments begin with the evaluation of hazardous situations that have the potential to cause harm. The harm could be to patients or to a downstream operation or output in the manufacturing process. Figure 1 provides a summary of how these layers are assembled into a full risk profile.
Figure 1. Progressive risk assessment model.

- Criticality is defined as the product of the Severity of Harm and the Likelihood of Harm. In some scenarios and especially with Critical Quality Attributes, there is an alternate definition used where the Criticality is the product of the Impact and the Uncertainty around the Impact. In this case, Uncertainty refers to how well the Impact is understood and whether the defined Impact is scientifically justified by direct data. We will explore Uncertainty in more detail later. In ISO 14971 terms, Criticality equals the product of the Severity of Harm (S) * the Probability of the Hazardous Situation leading to Harm (P2).
- Process Risk is defined as the product of Criticality and Occurrence. In QbDVision we also refer to Occurrence as Capability Risk. In a manufacturing process, if one thinks of occurrence as the probability that a process variable will be out-of-specification, then this is directly related to the process capability which can be statistically calculated from manufacturing data. Lower process capability means higher Capability Risk or probability of being out-of-specification. In ISO 14971 terms, Occurrence/Capability Risk is the Probability of the Hazardous Situation Occurring (P1) and the Risk as defined in this standard is equivalent to the Process Risk defined in QbDVision.
- Risk Priority Number is an overall risk metric that adds in the layer of Detectability Risk. Detectability is the concept that the hazardous situation can be detected before there is the incidence of harm. Detectability is often defined in terms of when the hazardous situation is detected relative to when the harm can occur. As an example, consider the pH of a drug formulation which can affect aggregation subsequently leading to cytotoxicity In the patient. If a pH out-of-specification can be detected at the time of manufacturing the bulk formulation, then the Detectability Risk is much lower than if it cannot be detected until testing at release. In QbDVision, RPN is calculated as the product of Process Risk and Detectability Risk which follows the general definition of Severity (S) * Occurrence (O) * Detectability (D). ISO 14971 does not include the concept of detectability in the calculation of risk or the idea of the RPN.
Figure 2 provides a graphical comparison of the QbDVision risk assessment structure with the ISO 14971 structure to demonstrate their alignment. Even though ISO 14971 does not include the concepts of detectability and RPN, they are included in figure 2 to allow comparison across all layers of risk.
Figure 2. Mathematical comparison of the risk models of ISO 14971 and QbDVision Risk Assessment.

Finally, it is important to note that Criticality is driven by Impact (Severity) and Uncertainty (Likelihood) and is not improved by reduction in Capability Risk (Occurrence) or Detectability Risk. Better process capability which reduces the probability of an attribute or parameter being outside of the acceptable range as demonstrated by manufacturing data, will reduce Process Risk. Similarly, earlier or more sensitive detection capability will reduce Detectability Risk and drive lower overall risk. With this structure, an attribute or parameter can be Critical but have a low overall risk with robust process capability and control. This is the methodology used to define your design space where these attributes and parameters can vary as long as they are within predefined limits without impacting critical quality attributes (CQAs).
Pretty simple, right?
Now that we have reviewed FMEA/FMECA, next week we’ll explore the impostors: uncertainty and detectability risk. While not as widely used, these two categories are important to creating clarity while assessing risk. We’ll discuss their usage and importance.
This post is part 3 of 7 in a series on practical risk management for pharmaceutical process development.


























































